Optical Pooled Screening Example
Before starting any full scale experiment, it is always recommended to explore a few tiles such that thresholds are set, segmentation is evaluated and in general have a better understanding of the data. This notebook walks you through some of the steps that you can use to do such a task
Load Experiment
The first step in any pipeline is to load the experiment. SCALLOPS can load single image with read_image, or an entire experiment (or a subset if requested) with read_experiment:
[1]:
from scallops.datasets import feldman_2019_small
from scallops.io import read_experiment
experiment_c_path = feldman_2019_small()
image_path = experiment_c_path / "input"
image_pattern = "{prefix}/{mag}X_c{t}-{experiment}-{t}_{well}_Tile-{tile}.{datatype}.tif" # Pattern that is followed by the images
In this particular example we will use several tiles of the public dataset experiment C. Its directory structure is (from the input directory):
├── 10X_c1-SBS-1
│ ├── 10X_c1-SBS-1_A1_Tile-102.sbs.tif
│ └── 10X_c1-SBS-1_A1_Tile-103.sbs.tif
├── 10X_c10-SBS-10
│ ├── 10X_c10-SBS-10_A1_Tile-102.sbs.tif
│ └── 10X_c10-SBS-10_A1_Tile-103.sbs.tif
├── 10X_c2-SBS-2
│ ├── 10X_c2-SBS-2_A1_Tile-102.sbs.tif
│ └── 10X_c2-SBS-2_A1_Tile-103.sbs.tif
├── 10X_c3-SBS-3
│ ├── 10X_c3-SBS-3_A1_Tile-102.sbs.tif
│ └── 10X_c3-SBS-3_A1_Tile-103.sbs.tif
├── 10X_c4-SBS-4
│ ├── 10X_c4-SBS-4_A1_Tile-102.sbs.tif
│ └── 10X_c4-SBS-4_A1_Tile-103.sbs.tif
├── 10X_c5-SBS-5
│ ├── 10X_c5-SBS-5_A1_Tile-102.sbs.tif
│ └── 10X_c5-SBS-5_A1_Tile-103.sbs.tif
├── 10X_c7-SBS-7
│ ├── 10X_c7-SBS-7_A1_Tile-102.sbs.tif
│ └── 10X_c7-SBS-7_A1_Tile-103.sbs.tif
├── 10X_c8-SBS-8
│ ├── 10X_c8-SBS-8_A1_Tile-102.sbs.tif
│ └── 10X_c8-SBS-8_A1_Tile-103.sbs.tif
└── 10X_c9-SBS-9
├── 10X_c9-SBS-9_A1_Tile-102.sbs.tif
└── 10X_c9-SBS-9_A1_Tile-103.sbs.tif
that is 9 cycles (missing cycle #6) of SBS data in two tiles of the well A1. The pattern that we have provided above ({prefix}/{mag}X_c{t}-{experiment}-{t}_{well}_Tile-{tile}.{datatype}.tif) contains:
prefix: this will store any parent directory name, e.g. 10X_c8-SBS-8, etc.
mag: Will store only the integer part of the magnification, 10 in this case.
t: Will store the t dimension. Since this is SBS data, it constitutes the cycle information.
experiment: The type of experiment.
well: from which well it comes from.
tile: which tile we are dealing with.
datatype: which kind of data it is. In this case, sbs.
All of these patterns can be arbitrary names that makes sense to you with the exception of the t, c, or z handles, as SCALLOPS will look for this name to store it appropriately.
Now that we know the structure of our data, let’s read it:
[2]:
iss_experiment = read_experiment(image_path, image_pattern, group_by=("well", "tile"))
print(iss_experiment)
print(iss_experiment.images.keys())
print(
read_experiment(
image_path, image_pattern, group_by=("well", "tile"), subset=["A1-102"]
)
) # If you want to subset your experiment
Experiment with 2 images and 0 labels
dict_keys(['A1-102', 'A1-103'])
Experiment with 1 image and 0 labels
Now we have our experiment loaded and ready to go. Now let’s access some images, and deal with the barcodes.
Load ISS Image and Barcodes
For some of the examples we will show you, we will use the image A1-102 as key. For experiment C the order of the bases was G, T, A C, and the dapi channel was capture first (index 0). All these parameters are important to do some of the processing of the data:
[3]:
import numpy as np
from scallops.io import read_barcodes
image_key = "A1-102" # Set the image key
dapi_channel = 0 # set a single dapi channel index
barcodes_path = experiment_c_path / "barcodes.csv"
iss_image = iss_experiment.images[image_key].max(
dim="z", keep_attrs=True
) # Get the image, and use the max z if any exist
iss_channels = np.delete(
np.arange(iss_image.sizes["c"]), dapi_channel
) # get the ISS only channels (remove dapi)
df_barcode = read_barcodes(
barcodes_path, iss_image.t.values - 1
) # Read the barcodes using the actual t indexes (missing cycle 6)
print("Image dimensions:", iss_image.sizes)
df_barcode.head()
Image dimensions: Frozen({'t': 9, 'c': 5, 'y': 1024, 'x': 1024})
[3]:
| barcode | sgRNA | gene_symbol | |
|---|---|---|---|
| 0 | CATTGACGC | CATTGCACGCCACAGCATTG | non-targeting |
| 1 | GAGTTCTAG | GAGTTTCTAGTAGTGGTAGG | non-targeting |
| 2 | AACGCGTCG | AACGCTGTCGTACGTGTATA | non-targeting |
| 3 | GCGGGCGGT | GCGGGGCGGTGACTTTCAAG | non-targeting |
| 4 | TTCTAGCCA | TTCTAAGCCACGTGTGGTAC | non-targeting |
Note that the barcodes contain all cycles except 6, as we expected. The read_barcodes is a utility simple function that also allows you to have a different name for the barcode by passing the barcode_column argument.
ISS Image
Now that we have a single image loaded into memory (iss_image), it is time to explore how it looks. There are a number of things that you might want to check, starting with what your data looks like in each channel. Luckily SCALLOPS’ plotting submodules will help with that. Let’s start with a montage plot of the channels with montage_plot:
[4]:
from scallops.visualize.composite import montage_plot
montage_plot(iss_image)
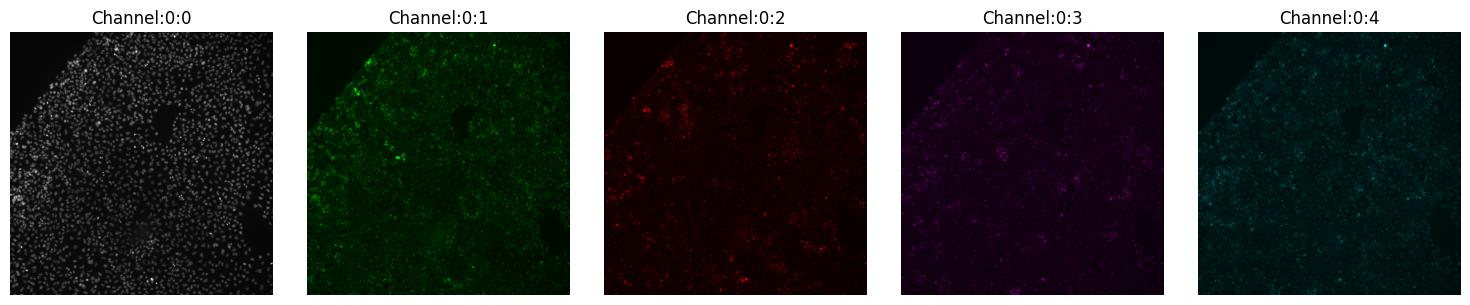
The figure above shows you every channel and you can make inferences about the quality, etc. However, it tells us very little about the alignment of the channels. During capture, channels and cycles can get misaligned, and that can create issues for downstream analyses.
Registration
The act of aligning channels and cycles is called registration. This is a crucial step, as it will be needed for base calling, etc. But before we go to the registration functions, let’s first verify how badly mis-aligned things are. SCALLOPS provides two ways to check this: imcomposite and diagnose_registration.
imcomposite
This shows a composite of all channels in a single image over a particular dimension. It is the most flexible and complete function for this task:
[5]:
from scallops.visualize.composite import imcomposite
imcomposite(iss_image.isel(c=dapi_channel), dim="t", figsize=(10, 10));
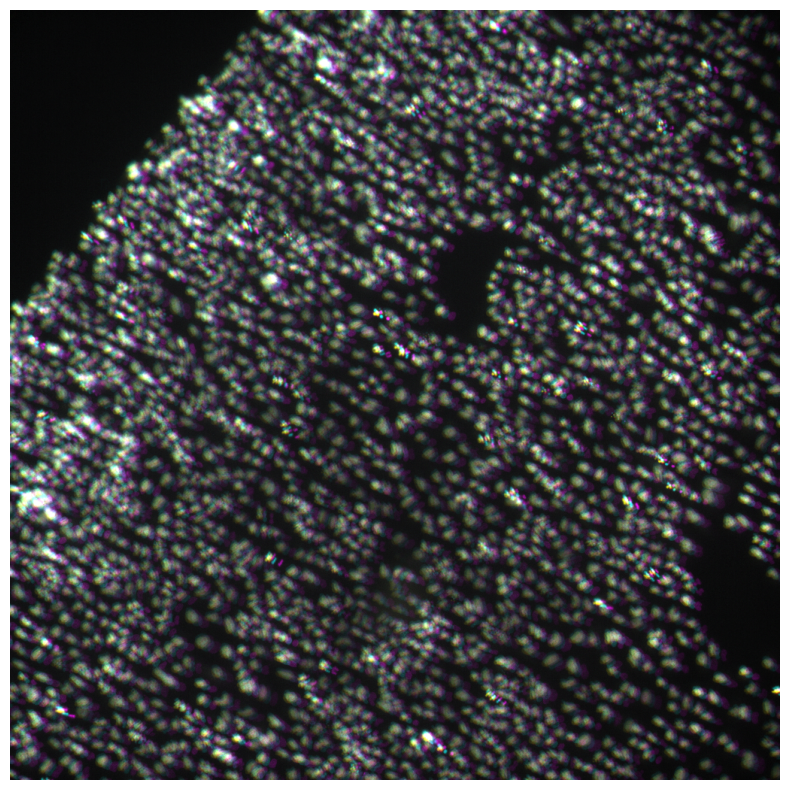
Here you can clearly see misalignment.
diagnose_registration
The other lightweight option is called diagnose_registration which is a simpler function. It has more limitations (only up to 6 images can be combined), but provides a bit more contrast than the imcomposite:
[6]:
import matplotlib.pyplot as plt
from scallops.visualize.registration import diagnose_registration
# Let's get some selectors
selectors = [
{"t": 1, "c": "Channel:0:0"}, # DAPI in first cycle
{"t": 1, "c": "Channel:0:1"}, # G in first cycle
{"t": 1, "c": "Channel:0:2"}, # T in first cycle
{"t": 1, "c": "Channel:0:3"}, # A in first cycle
{"t": 1, "c": "Channel:0:4"}, # C in first cycle
{"t": 2, "c": "Channel:0:0"}, # DAPI in second cycle
]
f, ax = plt.subplots(figsize=(10, 10))
axes = diagnose_registration(iss_image, *selectors, title="Before alignment")

The reason for the cap in the number of selectors is to keep the colormaps more dissimilar.
Both above cases showed us that the cycles and channels are not fully aligned, and we need to deal with this. This process is called registration. You can register an images with align_image:
[7]:
from scallops.registration.crosscorrelation import align_image
iss_image = align_image(
iss_image,
align_within_time_channels=iss_channels, # Ensure we align all channels within a cycle
align_between_time_channel=dapi_channel, # Ensure we align all cycles
filter_percentiles=[0, 90], # reduces potential noise during aligning (is optional)
)
imcomposite(iss_image.isel(c=dapi_channel), dim="t", figsize=(10, 10));
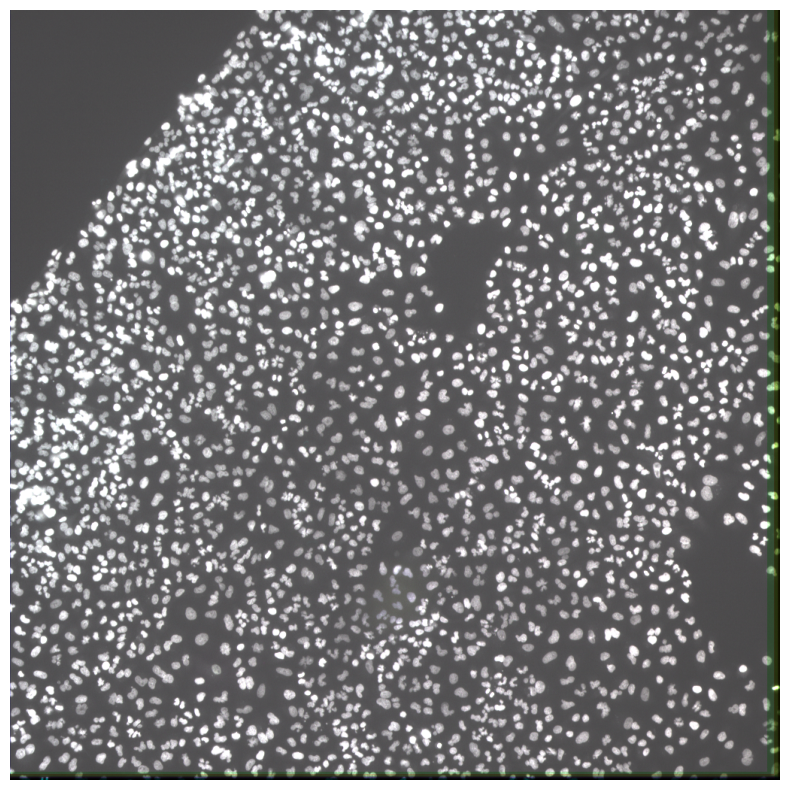
Now our ISS image is aligned.
Segmentation
The next step after registration is to segment the cellular compartments, starting with nuclei. SCALLOPS provides multiple algorithms for segmentation.
Segment Nuclei
Most OPS experiments start with segementing the nuclei. In fact most of our cellular segmentation options are seeded with the nuclear mask. For this tutorial we will focus on the stardist method, as we find it usually gives a good segmentation. That being said, the proper segmentation algorithm is very dataset dependent, so we encourage you to explore multiple algorithms. In SCALLOPS, stardist nuclear segmentation is called segment_nuclei_stardist.
[8]:
from scallops.segmentation.stardist import segment_nuclei_stardist
all_nuclei = segment_nuclei_stardist(image=iss_image, nuclei_channel=dapi_channel)
print(f"Nuclei, {len(np.unique(all_nuclei)) - 1:,}")
bioimageio_utils.py (2): pkg_resources is deprecated as an API. See https://setuptools.pypa.io/en/latest/pkg_resources.html. The pkg_resources package is slated for removal as early as 2025-11-30. Refrain from using this package or pin to Setuptools<81.
tf2onnx_lib.py (8): In the future `np.object` will be defined as the corresponding NumPy scalar.
Nuclei, 2,968
After acquiring the nuclei mask we would like to:
Visualize the mask
Check some geometric features
These two steps will help you verify and filter potential artifact of bad segmented areas, and define thresholds. For the first one we can use imcomposite again:
[9]:
imcomposite(
iss_image.isel(c=dapi_channel), dim="t", figsize=(10, 10), labels_contour=all_nuclei
);

Now lets look at the distribution of nuclei areas and set some thresholds. To extract this feature we will be using skimage.measure.regionprops_table:
[10]:
from skimage.measure import regionprops_table
nuclei_props = regionprops_table(all_nuclei, properties=["area"])
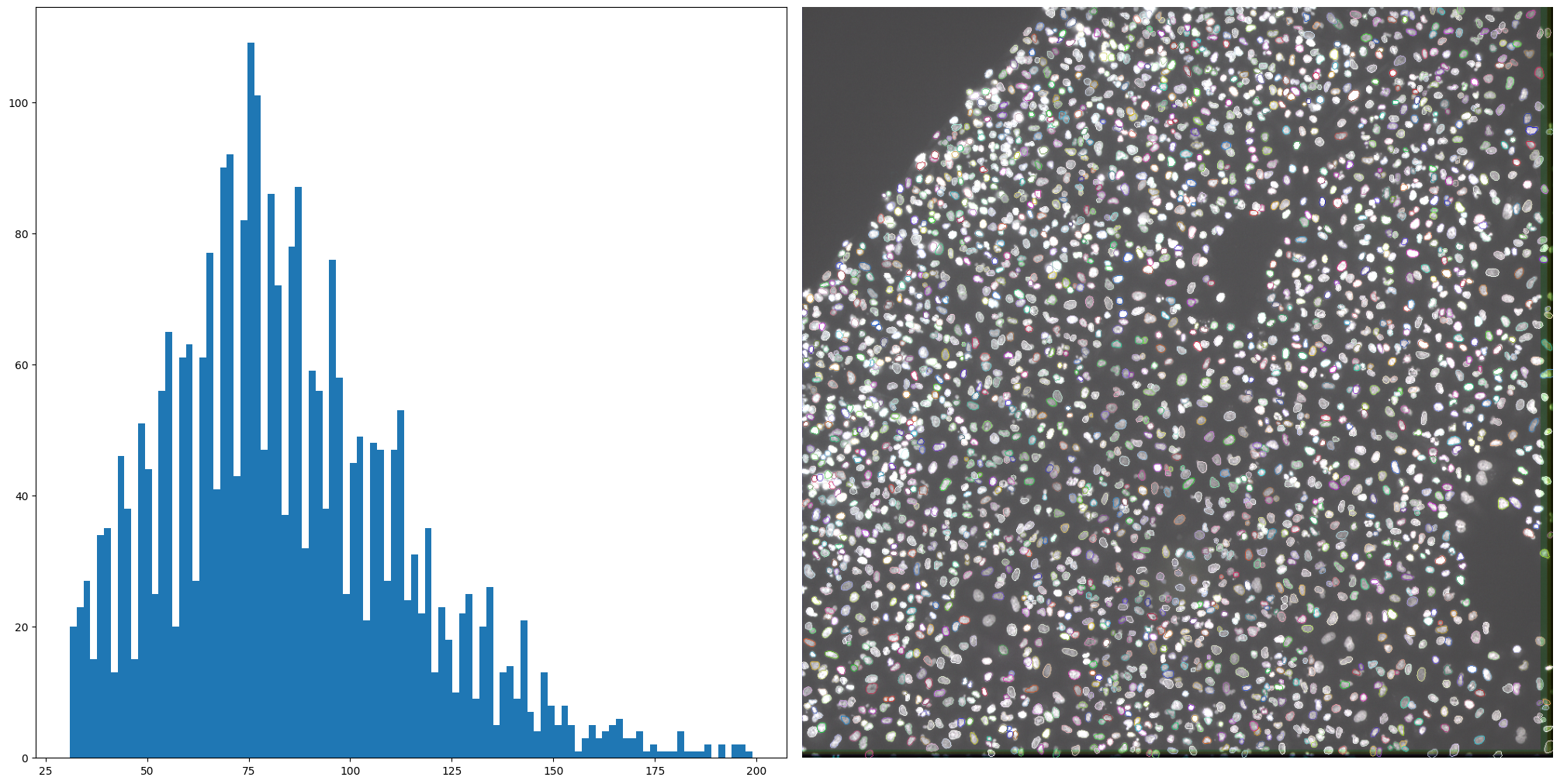
plt.hist(nuclei_props["area"], bins=100);
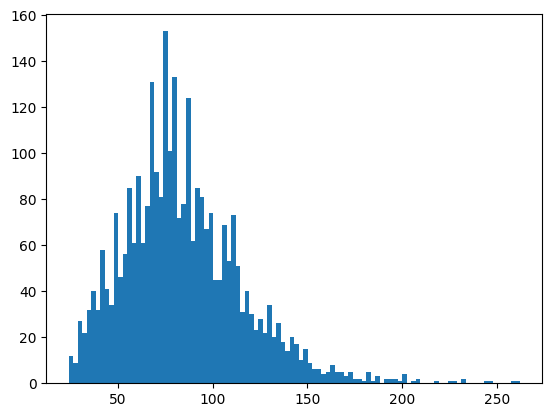
The distribution shows us a long tail, which is something that you would want to be inspected. From the visualization we can also see that there are a set of nuclei that seem too small. For the purposes of this example, we will assume that objects smaller than 30 and bigger than 200 are either artifacts, cells in mitotic state, or in general something we would like to filter out. SCALLOPS has a utility function filter_labels_by_area:
[11]:
from scallops.segmentation.util import remove_labels_by_area
nuclei_area_min = 30
nuclei_area_max = 200
nuclei = remove_labels_by_area(all_nuclei, nuclei_area_min, nuclei_area_max)
nuclei_props = regionprops_table(nuclei, properties=["area"])
f, axs = plt.subplots(ncols=2, figsize=(20, 10), layout="constrained")
axs[0].hist(nuclei_props["area"], bins=100)
imcomposite(iss_image.isel(c=dapi_channel), dim="t", labels_contour=nuclei, ax=axs[1]);
Segment cells
Now that we nuclei, we segment the cells. In some datasets, we found that given good nuclei seeds, watershed does an good job in segmenting the cells if the cell staining channel is well saturated. For this example we will be using segment_cells_watershed. In order to so, however, we do need to provide a threshold. There are a number of automatic algorithms available in this function (see API), but for simplicity we will pass a single threshold value. To do this, we need to explore the
distribution of the intensities, including and excluding nuclei. Since our data so far is ISS, we do not have a cytosolic channel (such as ConA for example). In these cases, we can create an average mask by using SCALLOPS’ function cyto_channel_summary:
[12]:
from scallops.segmentation.util import cyto_channel_summary
cell_mask = cyto_channel_summary(
iss_image.isel(t=0, c=iss_channels)
) # get an average mask for cells
f, ax = plt.subplots(figsize=(6, 6))
cell_mask_clipped = cell_mask.clip(
max=cell_mask.quantile(0.98)
) # Avoid really bright spots
in_nuc = cell_mask_clipped.data[nuclei > 0].flatten() # Get values in nuclei
out_nucl = cell_mask_clipped.data[nuclei == 0].flatten() # Get values outside nuclei
ax.boxplot([in_nuc, out_nucl], tick_labels=["In Nuclei", "Not in Nuclei"])
ax.set_ylabel("Intensity in Cell Mask");
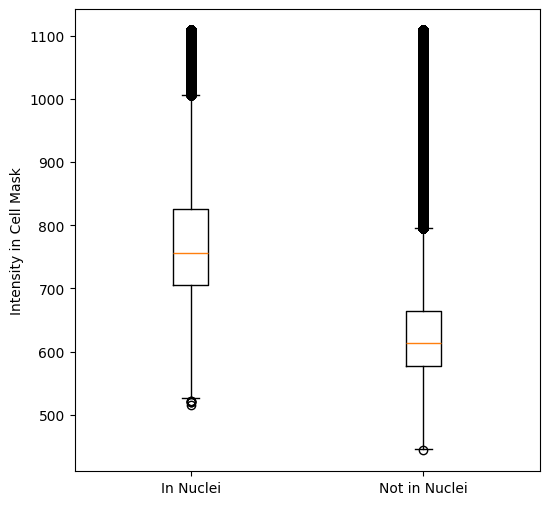
Judging by the above plot we set the cell threshold to 650:
[13]:
from scallops.segmentation.watershed import segment_cells_watershed
cell_segmentation_threshold = 650 # Set the threshold
all_cells, _ = segment_cells_watershed(
iss_image, nuclei, threshold=cell_segmentation_threshold
) # segment the cells
imcomposite(
iss_image.isel(c=dapi_channel),
dim="t",
labels=nuclei, # visualize nuclei solid
labels_contour=all_cells, # visualize cells as boundaries
figsize=(10, 10),
)
plt.title(f"Cells, {len(np.unique(all_cells)) - 1:,}");

We can see a pretty good segmentation (despite some holes and lack of definition here and there) given that there is no cytosolic mask. Let us assume that this segmentation is good, and lets clean it up a little. Similar to the nuclei, we can visualize and inspect the cellular area, and then filter based on set thresholds:
[14]:
cell_props = regionprops_table(all_cells, properties=["area"])
plt.hist(cell_props["area"], bins=100);
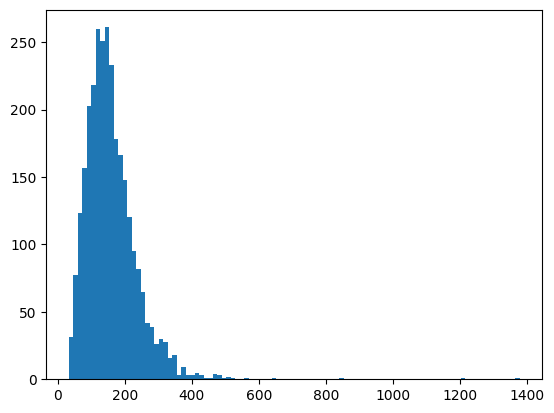
Now we can set the minimum and maximum areas allowed to 40 and 500:
[15]:
cell_area_min = 40
cell_area_max = 500
cells = remove_labels_by_area(all_cells, cell_area_min, cell_area_max)
cell_props = regionprops_table(cells, properties=["area"])
fig, axes = plt.subplots(1, 2, figsize=(10, 5), layout="constrained")
axes[0].set_title("Cell Area")
axes[0].hist(cell_props["area"], bins=100)
axes[1].set_title(f"Cells, {len(np.unique(cells)) - 1:,}")
imcomposite(image=iss_image.isel(c=dapi_channel), dim="t", labels=cells, ax=axes[1])
axes[1].axis("off");
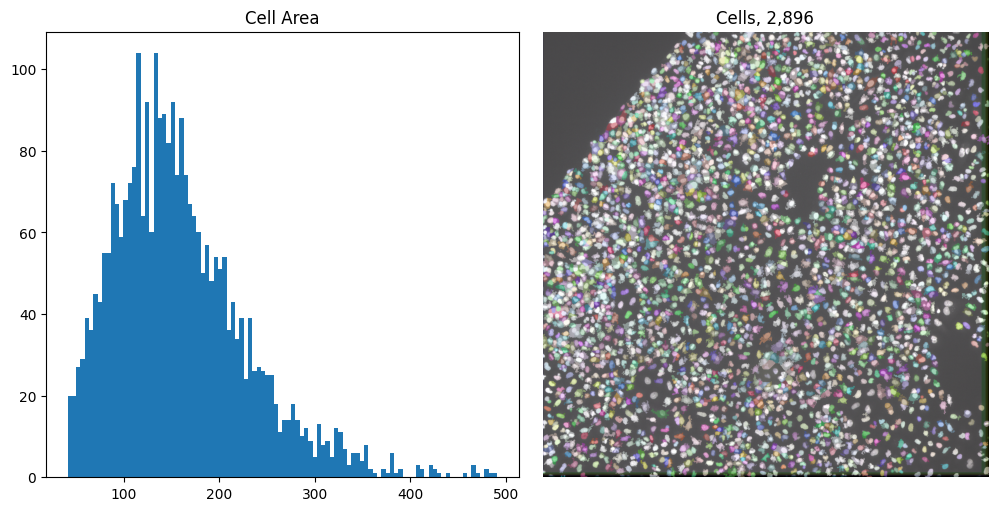
Now we have both cells and nuclei segmented, we can move to barcode calling.
Barcode calling
In OPS to call the barcodes we need to:
Find peaks: Identify the intensity peaks that are shared across all ISS channels
Filter peaks: Identify false positive and filter them
Cross-talk correction: Compute the cross-talk and corrected
Call Bases: Based on the max corrected intensities in each cycle, call the bases
Call reads: Call reads by calling the base with the highest intensity
Find Peaks
Finding the peaks requires to denoise the image with Laplacian of Gaussian (LoG) transform, followed by computing the standard deviation across sequencing channels, to identify where peaks are. This can be done with transform_log, std, and find_peaks, respectively:
[16]:
from scallops.spots import find_peaks, std, transform_log
loged = transform_log(iss_image.isel(c=iss_channels))
std_arr = std(loged)
peaks = find_peaks(std_arr)
bases_array = find_peaks(std_arr)
Call Bases
To call bases we need to compute the maximum filter over the LoG transform using the max_filter function followed by the calling function peaks_to_bases:
[17]:
from scallops.reads import peaks_to_bases
from scallops.spots import max_filter
maxed = max_filter(loged)
bases_array = peaks_to_bases(maxed=maxed, peaks=peaks, labels=cells)
bases_array.sizes
[17]:
Frozen({'read': 10557, 't': 9, 'c': 4})
Now we have all unfiltered peaks. We divide the unfiltered peaks into high quality and low quality peaks by computing the probability of the bases in the read at the peak location. We choose the peak threshold with the maximimum F0.5 score using . peak_thresholds_from_bases
[18]:
from scallops.spots import peak_thresholds_from_bases
threshold_peaks_crosstalk_correction_df = peak_thresholds_from_bases(
bases_array=bases_array
)
threshold_peaks_crosstalk_correction = threshold_peaks_crosstalk_correction_df.iloc[0][
"threshold"
]
f"Crosstalk peak threshold: {threshold_peaks_crosstalk_correction}"
[18]:
'Crosstalk peak threshold: 103.40227203832168'
Cross-talk correction
Now we can visualize the cross-talk with pairwise_channel_scatter_plot:
[19]:
from scallops.visualize.crosstalk import pairwise_channel_scatter_plot
fig, _ = pairwise_channel_scatter_plot(
bases_array.where(
bases_array.peak > threshold_peaks_crosstalk_correction, drop=True
)
)
fig.suptitle("Uncorrected Intensity (In Cells)")
fig.tight_layout()
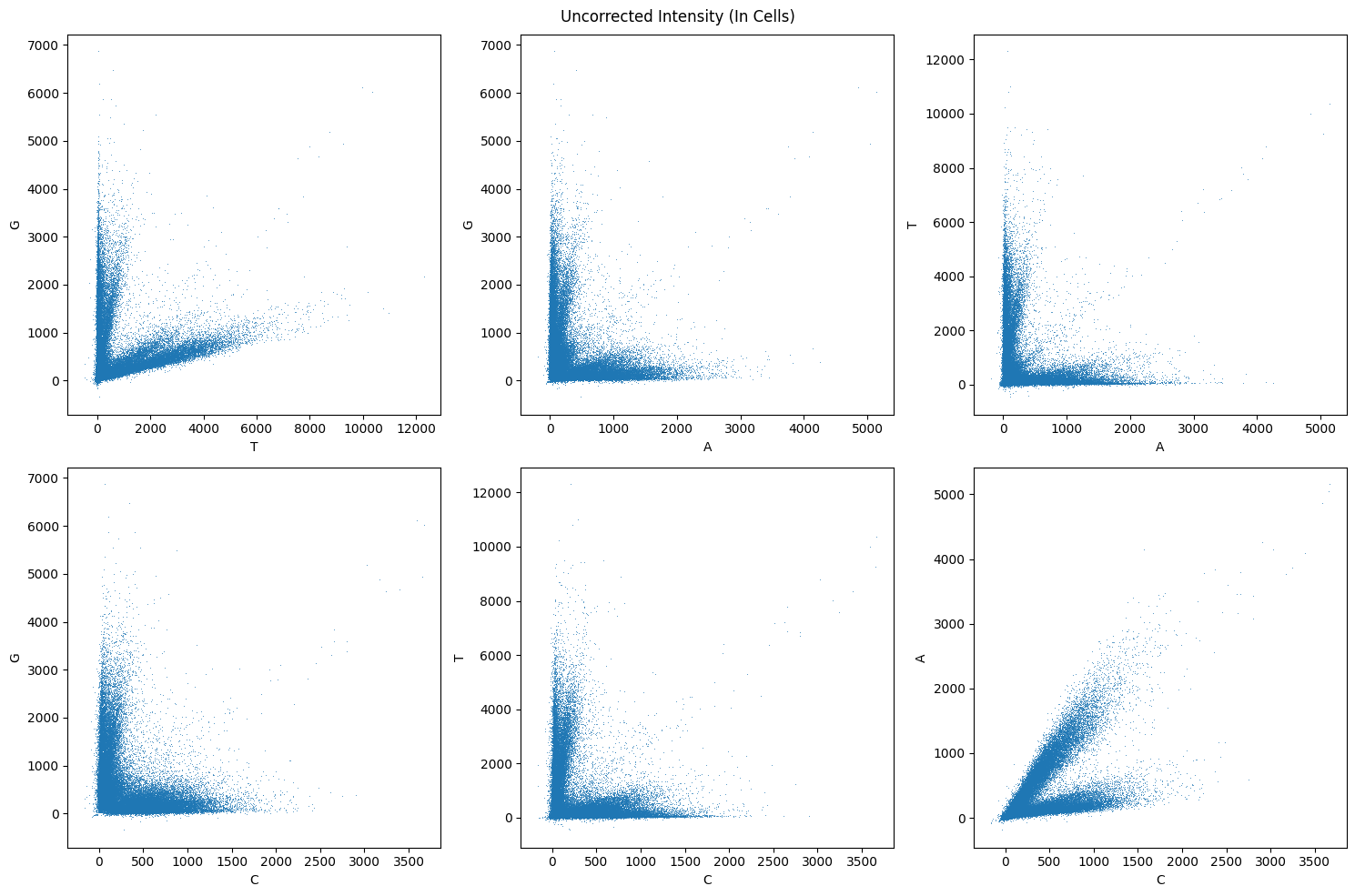
We can see that there is some cross-talk, especially betwen A and C and T and G. Now we need to compute the cross-talk matrix with channel_crosstalk_matrix, correct the cross-talk with apply_channel_crosstalk_matrix, and again visualize the result of the correction.
[20]:
from scallops.reads import apply_channel_crosstalk_matrix, channel_crosstalk_matrix
w = channel_crosstalk_matrix(
bases_array.where(
bases_array.peak > threshold_peaks_crosstalk_correction, drop=True
)
)
corrected_bases_array = apply_channel_crosstalk_matrix(bases_array, w)
fig, _ = pairwise_channel_scatter_plot(corrected_bases_array)
fig.suptitle("Corrected Intensity (In Cells)")
fig.tight_layout()
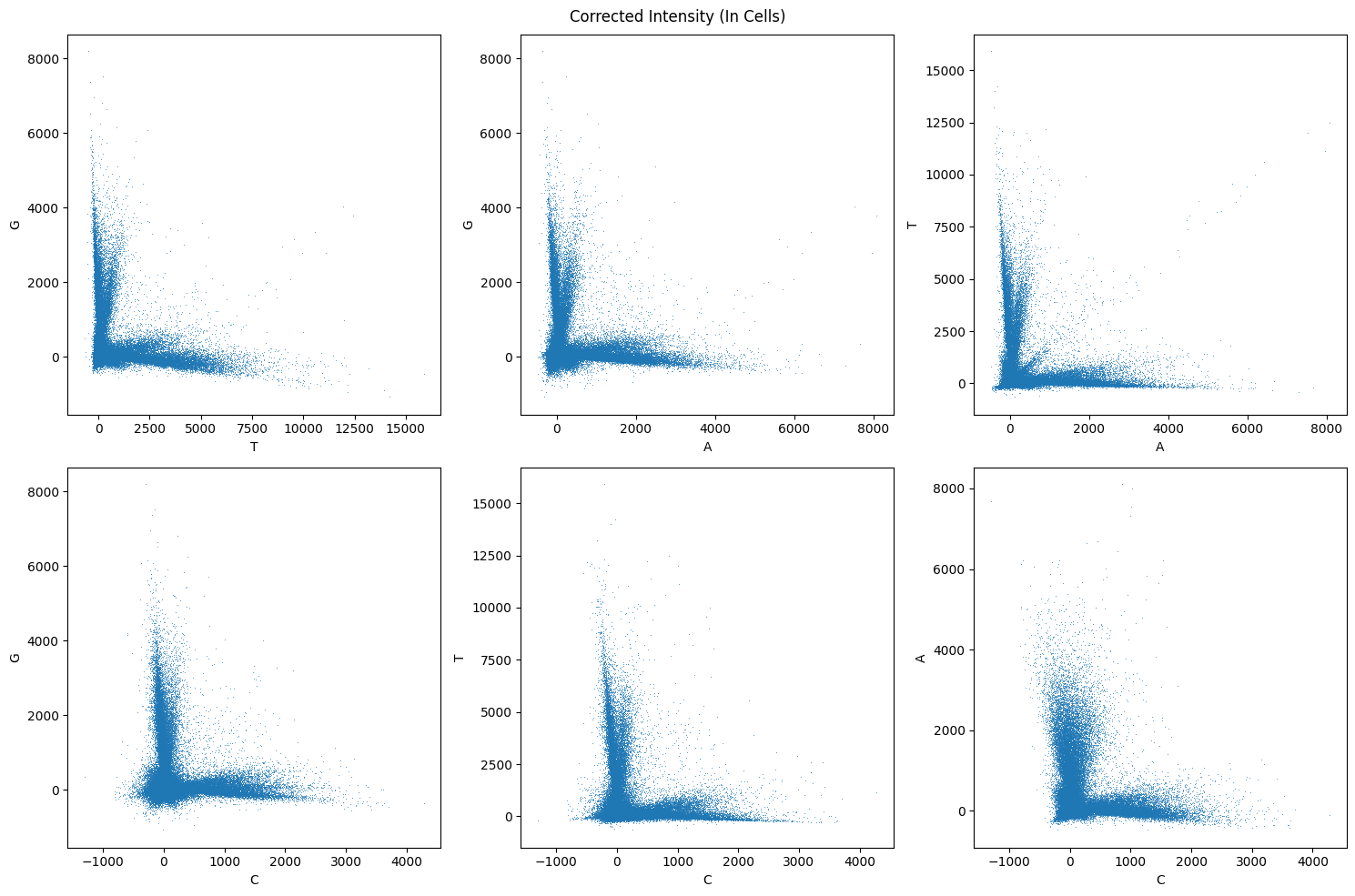
Call Reads
Now that we have a corrected set of base intensities, we can call the reads using SCALLOPS’ decode-max function, and we can see some descriptive read statistics with read_statistics:
[21]:
from scallops.reads import decode_max, read_statistics
from scallops.spots import peak_thresholds_from_reads
df_reads = decode_max(corrected_bases_array, barcodes=df_barcode)
peak_thresholds_lower_df = peak_thresholds_from_reads(df_reads.query("barcode_match"))
threshold_peaks = peak_thresholds_lower_df.iloc[0]["threshold"]
f"Lower peak threshold: {threshold_peaks:.2f}"
[21]:
'Lower peak threshold: 49.46'
[22]:
df_reads = df_reads.query(f"peak>{threshold_peaks}")
read_statistics(df_reads)
[22]:
{'mapped_reads': 5981,
'number_of_reads': 7632,
'mapping_rate': 0.7836740041928721,
'mapping_rate_within_labels': 0.7836740041928721,
'mapped_reads_within_labels': 5981,
'average_reads_per_label': 3.0974025974025974,
'average_mapped_reads_per_label': 2.427353896103896,
'number_of_unique_barcodes_in_labels': 825,
'mean_barcode_count_in_labels': 7.24969696969697,
'labels_with_reads': 2464,
'labels_with_mapped_reads': 2202}
Visualize Single Base Calling Errors
After base calling we want to visualize the biases and mismatches across our bases. We can obtain the summary of the mismatches using SCALLOPS’ summarize_base_call_mismatches, and then visualize with base_call_mismatches_heatmap:
[23]:
from scallops.reads import summarize_base_call_mismatches
from scallops.visualize.heatmap import base_call_mismatches_heatmap
base_call_mismatches_df = summarize_base_call_mismatches(df_reads, df_barcode)
display(base_call_mismatches_df.head())
base_call_mismatches_heatmap(base_call_mismatches_df);
| called_base | whitelist_base | read_position | count | fraction | |
|---|---|---|---|---|---|
| 0 | A | C | 0 | 2 | 0.003868 |
| 1 | A | C | 1 | 2 | 0.003868 |
| 2 | A | C | 2 | 2 | 0.003868 |
| 3 | A | C | 3 | 2 | 0.003868 |
| 4 | A | C | 4 | 8 | 0.015474 |
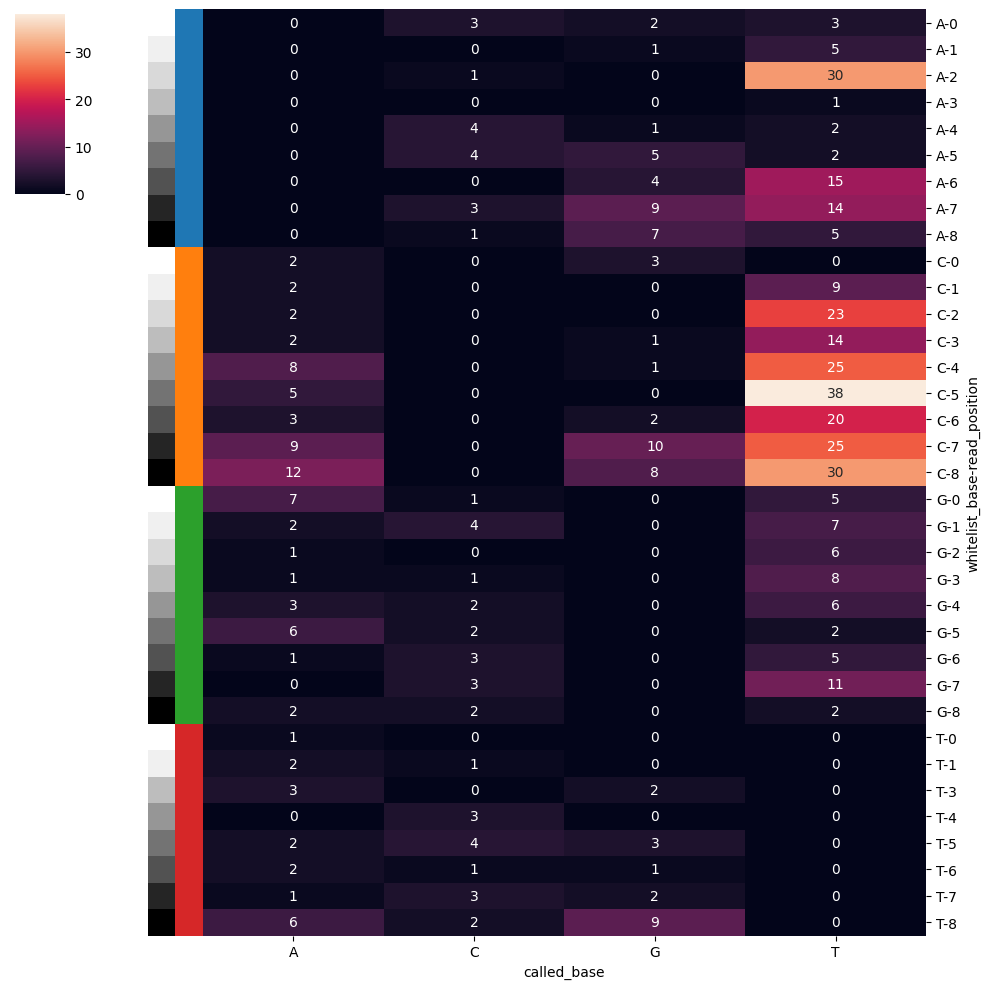
Correct Reads With One Mismatch
The figure above showed us that there are mismatches from our expected barcode list. We can try fliping bases to match the whitelist. This can be done by computing the hamming distance and restricting the flipping to a desired number of mismatches you want fixed. This can easily be achieved by SCALLOPS’ correct_mismatches function:
[24]:
from scallops.reads import correct_mismatches
df_reads = correct_mismatches(df_reads, df_barcode)
read_statistics(df_reads)
[24]:
{'mapped_reads': 6498,
'number_of_reads': 7632,
'mapping_rate': 0.8514150943396226,
'mapping_rate_within_labels': 0.8514150943396226,
'mapped_reads_within_labels': 6498,
'average_reads_per_label': 3.0974025974025974,
'average_mapped_reads_per_label': 2.637175324675325,
'number_of_unique_barcodes_in_labels': 949,
'mean_barcode_count_in_labels': 6.847207586933615,
'labels_with_reads': 2464,
'labels_with_mapped_reads': 2297}
[25]:
df_reads.head()
[25]:
| y | x | sigma | peak | label | read | barcode | Q | Q_mean | Q_min | barcode_match | mismatches | closest_match | mismatches2 | closest_match2 | barcode_uncorrected | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 19 | 5 | 756 | 1 | 1189.094209 | 924 | 19 | AAGCCAATT | [60.0, 60.0, 60.0, 60.0, 60.0, 60.0, 60.0, 60.... | 60.0 | 60.0 | True | NaN | NaN | NaN | NaN | NaN |
| 20 | 5 | 791 | 1 | 488.943090 | 2258 | 20 | AGCATGCCA | [60.0, 60.0, 60.0, 60.0, 60.0, 60.0, 60.0, 60.... | 60.0 | 60.0 | True | NaN | NaN | NaN | NaN | NaN |
| 21 | 5 | 796 | 1 | 372.566588 | 2258 | 21 | AGCATGCCA | [60.0, 60.0, 60.0, 60.0, 60.0, 60.0, 60.0, 60.... | 60.0 | 60.0 | True | NaN | NaN | NaN | NaN | NaN |
| 22 | 5 | 809 | 1 | 187.319520 | 2258 | 22 | AAGAGTTCT | [60.0, 60.0, 60.0, 60.0, 60.0, 60.0, 60.0, 60.... | 60.0 | 60.0 | True | NaN | NaN | NaN | NaN | NaN |
| 39 | 6 | 481 | 1 | 58.776617 | 1506 | 39 | AAGGGGTCT | [60.0, 60.0, 60.0, 60.0, 60.0, 60.0, 60.0, 60.... | 60.0 | 60.0 | False | 1.0 | AAGGCGTCT | 1.0 | AAGGGCTCT | NaN |
Our overall mapping rate is now higher and we also find more cells with mapped reads than before the correction.
Interactive visualization of Base Calls
We can use Napari to interactively visualize the base calls and the underlying data with SCALLOPS’ add_bases:
[26]:
import napari
from scallops.visualize.napari import add_bases
viewer = napari.Viewer()
viewer.add_image(
iss_image.squeeze(),
channel_axis=1,
visible=[False, True, True, True, True],
name=["DAPI"] + ["G", "T", "A", "C"],
)
add_bases(viewer, df_reads)
viewer.add_labels(
cells,
name="cells",
colormap=napari.utils.DirectLabelColormap(color_dict={None: (1, 1, 1, 0.5)}),
).contour = 1
The above code should open a new window with napari, and the bases should be in the left-hand side panel. A zoom in should look like this:

Assign Barcodes To Cells
So far we have successfully called the barcodes, but we still need to assign the barcode to each label. This can be achieved by calling assign_barcodes_to_labels:
[27]:
from scallops.reads import assign_barcodes_to_labels
df_cells = assign_barcodes_to_labels(df_reads).set_index("label")
df_cells
[27]:
| mismatch | barcode_Q_mean | barcode_Q_min | barcode_peak | barcode_count | barcode_0 | barcode_Q_mean_0 | barcode_Q_min_0 | barcode_peak_0 | barcode_count_0 | barcode_Q_0 | barcode_1 | barcode_Q_mean_1 | barcode_Q_min_1 | barcode_peak_1 | barcode_count_1 | barcode_Q_1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| label | |||||||||||||||||
| 924 | False | 60.000000 | 60.000000 | 1189.094209 | 1 | AAGCCAATT | 60.000000 | 60.000000 | 1189.094209 | 1 | [60.0, 60.0, 60.0, 60.0, 60.0, 60.0, 60.0, 60.... | 0.000000 | 0.000000 | NaN | 0 | None | |
| 2258 | False | 300.000000 | 300.000000 | 1801.712283 | 5 | AGCATGCCA | 180.000000 | 180.000000 | 1479.391942 | 3 | [180.0, 180.0, 180.0, 180.0, 180.0, 180.0, 180... | AAGAGTTCT | 60.000000 | 60.000000 | 187.319520 | 1 | [60.0, 60.0, 60.0, 60.0, 60.0, 60.0, 60.0, 60.... |
| 1506 | True | 60.000000 | 60.000000 | 58.776617 | 1 | AAGGGGTCT | 60.000000 | 60.000000 | 58.776617 | 1 | [60.0, 60.0, 60.0, 60.0, 60.0, 60.0, 60.0, 60.... | 0.000000 | 0.000000 | NaN | 0 | None | |
| 2900 | False | 120.000000 | 120.000000 | 1040.681147 | 2 | AAGCCAATT | 120.000000 | 120.000000 | 1040.681147 | 2 | [120.0, 120.0, 120.0, 120.0, 120.0, 120.0, 120... | 0.000000 | 0.000000 | NaN | 0 | None | |
| 2664 | False | 238.460434 | 226.906937 | 1075.447078 | 4 | TCCATTGCA | 238.460434 | 226.906937 | 1075.447078 | 4 | [240.0, 226.90694, 240.0, 240.0, 240.0, 240.0,... | 0.000000 | 0.000000 | NaN | 0 | None | |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 1291 | True | 106.944305 | 2.498775 | 1709.110210 | 2 | TTTGGGCAG | 53.472153 | 1.249387 | 1355.472936 | 1 | [60.0, 60.0, 60.0, 60.0, 60.0, 60.0, 60.0, 60.... | GGGGTGCAG | 53.472153 | 1.249387 | 353.637274 | 1 | [60.0, 60.0, 60.0, 60.0, 60.0, 60.0, 60.0, 60.... |
| 2826 | True | 53.472153 | 1.249387 | 87.979374 | 1 | ACAACCCAG | 53.472153 | 1.249387 | 87.979374 | 1 | [60.0, 60.0, 60.0, 60.0, 60.0, 60.0, 60.0, 60.... | 0.000000 | 0.000000 | NaN | 0 | None | |
| 2584 | True | 47.288986 | 1.249387 | 232.257161 | 1 | GCCATGCTG | 47.288986 | 1.249387 | 232.257161 | 1 | [60.0, 60.0, 60.0, 60.0, 60.0, 60.0, 4.35147, ... | 0.000000 | 0.000000 | NaN | 0 | None | |
| 2539 | True | 47.441696 | 1.249387 | 458.368261 | 1 | GGCAGGCTG | 47.441696 | 1.249387 | 458.368261 | 1 | [60.0, 60.0, 60.0, 60.0, 60.0, 60.0, 5.725899,... | 0.000000 | 0.000000 | NaN | 0 | None | |
| 2768 | True | 47.338661 | 1.249387 | 54.133295 | 1 | ATTTTTCTG | 47.338661 | 1.249387 | 54.133295 | 1 | [58.82297, 60.0, 60.0, 60.0, 60.0, 60.0, 5.975... | 0.000000 | 0.000000 | NaN | 0 | None |
2464 rows × 17 columns
Now the label ID is in the index of df_cells.
Extracting phenotype information
Now that we have our barcode called, we can extract the phenotypes. These steps are independent of the ISS steps and can be run in parallel up to the merging step.
Loading phenotypic data
The loading of the pheno data proceeds in the same fashion as the ISS data:
[28]:
phenotype_image_path = experiment_c_path / "10X_c0-DAPI-p65ab"
phenotype_image_pattern = (
"{mag}X_c{t}-{experiment}-{t}_{well}_Tile-{tile}.{datatype}.tif"
)
phenotype_experiment = read_experiment(phenotype_image_path, phenotype_image_pattern)
print(phenotype_experiment)
print(phenotype_experiment.images.keys())
Experiment with 2 images and 0 labels
dict_keys(['A1-102', 'A1-103'])
[29]:
phenotype_experiment.images[image_key].sizes # using the same image key as in ISS
[29]:
Frozen({'t': 1, 'c': 2, 'z': 1, 'y': 1024, 'x': 1024})
Note that this image contains only one “cycle” or timepoint, and only 2 channels of phenotyping. The channels correspond to DAPI and to the NF-κB protein expression.
Alignment with ISS
Since our goal is to estimate features and match it with the ISS data, is important to have a reliable registration between the two modalities (ISS and phenotype). In this case both modalities are in 10X and the alignment between them is not too off, therefore, we can use the cross correlation alignment again. Note that SCALLOPS also provides ITK registration.
[30]:
from scallops.registration.crosscorrelation import ( # note that this is a different function than align_image
align_images,
)
pheno_image = phenotype_experiment.images[image_key].max(dim="z", keep_attrs=True)
pheno_aligned = align_images(iss_image.isel(t=0), pheno_image.isel(t=0))
pheno_aligned.sizes
[30]:
Frozen({'c': 2, 'y': 1024, 'x': 1024})
We can check how good the alignment is by either plotting a phenotype composite with the ISS labels or we can merge the two images and visualize the composite:
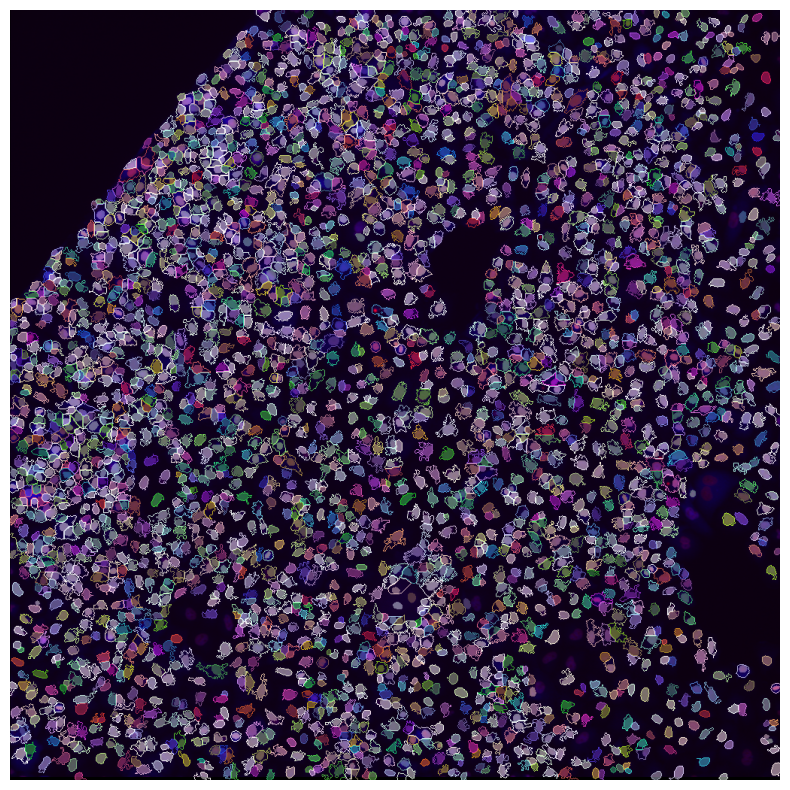
[31]:
imcomposite(pheno_aligned, labels=nuclei, labels_contour=cells, figsize=(10, 10));
[32]:
import xarray as xr
combined = xr.concat(
[iss_image, pheno_aligned.expand_dims(["t"])], dim="c", join="outer"
)
imcomposite(combined.isel(c=dapi_channel), dim="t", figsize=(10, 10));
Compute Features
To compute the features you need to pass one of the features you want with the parameters for said features. In this example we will get the area, eccentricity and the correlation between DAPI (channel index 0) and NF-κB (channel index 1):
[33]:
from scallops.features.find_objects import find_objects
from scallops.features.generate import label_features
objects_df = find_objects(cells)
phenotype_results = label_features(
objects_df=objects_df,
intensity_image=pheno_aligned.transpose(
*("y", "x", "c")
).data, # data needs to be transposed so that channel is last
label_image=cells, # You can pass any segmentation label
features=["intensity_*", "size-shape"],
)
df_phenotype = objects_df.join(phenotype_results)
df_phenotype
measureobjectsizeshape.py (637): divide by zero encountered in divide
zernike.py (181): invalid value encountered in divide
zernike.py (184): invalid value encountered in divide
[33]:
| AreaShape_BoundingBoxMinimum_Y | AreaShape_BoundingBoxMaximum_Y | AreaShape_BoundingBoxMinimum_X | AreaShape_BoundingBoxMaximum_X | AreaShape_Center_Y | AreaShape_Center_X | AreaShape_Area | AreaShape_BoundingBoxArea | AreaShape_ConvexArea | AreaShape_EquivalentDiameter | ... | Intensity_IntegratedIntensityEdge_Channel0 | Intensity_MeanIntensityEdge_Channel0 | Intensity_StdIntensityEdge_Channel0 | Intensity_MinIntensityEdge_Channel0 | Intensity_MaxIntensityEdge_Channel0 | Intensity_IntegratedIntensityEdge_Channel1 | Intensity_MeanIntensityEdge_Channel1 | Intensity_StdIntensityEdge_Channel1 | Intensity_MinIntensityEdge_Channel1 | Intensity_MaxIntensityEdge_Channel1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 117 | 136 | 698 | 718 | 125.938525 | 708.340164 | 244.0 | 380.0 | 261.0 | 17.625846 | ... | 0.515236 | 0.010103 | 0.003856 | 0.007797 | 0.026612 | 1.439216 | 0.028220 | 0.007859 | 0.019028 | 0.053559 |
| 2 | 418 | 440 | 355 | 378 | 427.787645 | 367.528958 | 259.0 | 506.0 | 352.0 | 18.159544 | ... | 0.871229 | 0.009470 | 0.002003 | 0.007767 | 0.017273 | 2.553872 | 0.027759 | 0.003378 | 0.021912 | 0.036515 |
| 3 | 264 | 280 | 491 | 510 | 271.485000 | 500.485000 | 200.0 | 304.0 | 223.0 | 15.957691 | ... | 0.490776 | 0.009438 | 0.001078 | 0.008225 | 0.012848 | 1.407126 | 0.027060 | 0.003307 | 0.020035 | 0.034806 |
| 4 | 208 | 228 | 663 | 676 | 217.015000 | 668.940000 | 200.0 | 260.0 | 221.0 | 15.957691 | ... | 0.606455 | 0.011891 | 0.004497 | 0.007965 | 0.027039 | 1.345937 | 0.026391 | 0.003446 | 0.019196 | 0.035111 |
| 5 | 509 | 534 | 896 | 916 | 521.124113 | 906.379433 | 282.0 | 500.0 | 322.0 | 18.948708 | ... | 0.674846 | 0.009780 | 0.002776 | 0.007721 | 0.021027 | 1.410269 | 0.020439 | 0.003635 | 0.013596 | 0.031800 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 2963 | 42 | 51 | 534 | 544 | 45.354167 | 539.437500 | 48.0 | 90.0 | 59.0 | 7.817640 | ... | 0.257374 | 0.010295 | 0.002209 | 0.008026 | 0.016495 | 0.550072 | 0.022003 | 0.003705 | 0.016495 | 0.031434 |
| 2964 | 816 | 823 | 627 | 642 | 819.115942 | 634.565217 | 69.0 | 105.0 | 76.0 | 9.373021 | ... | 0.358755 | 0.011958 | 0.003057 | 0.008164 | 0.017624 | 0.744167 | 0.024806 | 0.005798 | 0.014466 | 0.035080 |
| 2965 | 798 | 813 | 410 | 427 | 805.957265 | 418.205128 | 117.0 | 255.0 | 189.0 | 12.205287 | ... | 0.674815 | 0.011063 | 0.004951 | 0.007691 | 0.025071 | 1.199924 | 0.019671 | 0.004311 | 0.014527 | 0.032441 |
| 2967 | 259 | 270 | 175 | 185 | 263.964286 | 180.571429 | 56.0 | 110.0 | 75.0 | 8.444016 | ... | 0.391089 | 0.013036 | 0.003799 | 0.008179 | 0.020432 | 0.852430 | 0.028414 | 0.004930 | 0.018250 | 0.038239 |
| 2968 | 487 | 499 | 918 | 928 | 492.149254 | 922.970149 | 67.0 | 120.0 | 81.0 | 9.236182 | ... | 0.390738 | 0.013025 | 0.005218 | 0.008042 | 0.026551 | 0.798428 | 0.026614 | 0.006177 | 0.019364 | 0.039643 |
2896 rows × 148 columns
[34]:
import pandas as pd
from scallops.reads import merge_sbs_phenotype
merged_df = merge_sbs_phenotype(
df_labels=df_cells,
df_phenotype=df_phenotype,
df_barcode=pd.read_csv(barcodes_path),
sbs_cycles=iss_image.coords["t"].values,
)
merged_df
[34]:
| mismatch | barcode_Q_mean | barcode_Q_min | barcode_peak | barcode_count | barcode_0 | barcode_Q_mean_0 | barcode_Q_min_0 | barcode_peak_0 | barcode_count_0 | ... | Intensity_MinIntensityEdge_Channel1 | Intensity_MaxIntensityEdge_Channel1 | barcode | sgRNA | gene_symbol | duplicate_prefix | barcode_1_barcode_1 | sgRNA_1 | gene_symbol_1 | duplicate_prefix_1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | False | 360.0 | 360.0 | 3272.963489 | 6.0 | AAATGATCT | 360.0 | 360.0 | 3272.963489 | 6.0 | ... | 0.019028 | 0.053559 | AAATGGATCTATGTTCACAA | AAATGGATCTATGTTCACAA | FBXO32 | False | NaN | NaN | NaN | NaN |
| 2 | True | 180.0 | 180.0 | 1098.411341 | 3.0 | AGGATTTAG | 180.0 | 180.0 | 1098.411341 | 3.0 | ... | 0.021912 | 0.036515 | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| 3 | False | 60.0 | 60.0 | 268.992267 | 1.0 | GCTGTAGAC | 60.0 | 60.0 | 268.992267 | 1.0 | ... | 0.020035 | 0.034806 | GCTGTGAGACCCAGCACCTG | GCTGTGAGACCCAGCACCTG | DHX58 | False | NaN | NaN | NaN | NaN |
| 4 | False | 300.0 | 300.0 | 1994.343939 | 5.0 | GCTCTGCAG | 300.0 | 300.0 | 1994.343939 | 5.0 | ... | 0.019196 | 0.035111 | GCTCTTGCAGGAATTTCCAG | GCTCTTGCAGGAATTTCCAG | USP17L2 | False | NaN | NaN | NaN | NaN |
| 5 | False | 300.0 | 300.0 | 2196.703452 | 5.0 | ACTCAAAAC | 300.0 | 300.0 | 2196.703452 | 5.0 | ... | 0.013596 | 0.031800 | ACTCACAAACAACATTGCTG | ACTCACAAACAACATTGCTG | IL6R | False | NaN | NaN | NaN | NaN |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 2963 | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | ... | 0.016495 | 0.031434 | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| 2964 | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | ... | 0.014466 | 0.035080 | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| 2965 | False | 180.0 | 180.0 | 1285.524961 | 3.0 | CTCACTGCA | 180.0 | 180.0 | 1285.524961 | 3.0 | ... | 0.014527 | 0.032441 | CTCACCTGCAATTAGGTGGA | CTCACCTGCAATTAGGTGGA | UBE2E1 | False | NaN | NaN | NaN | NaN |
| 2967 | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | ... | 0.018250 | 0.038239 | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| 2968 | False | 60.0 | 60.0 | 64.093509 | 1.0 | GCACGGTGG | 60.0 | 60.0 | 64.093509 | 1.0 | ... | 0.019364 | 0.039643 | GCACGTGTGGCAGATCACGT | GCACGTGTGGCAGATCACGT | SPSB1 | False | NaN | NaN | NaN | NaN |
2896 rows × 173 columns
Post-processing results
After getting all the features and barcode results, we still need to process the data so it is clean and trustworthy.
Keep cells in which top barcode accounts for > 50% of aligned reads
The so-called 50% rule allows you to have more trust in your data. When you have multiple barcodes in a cell, you’d like that the top barcode represents more than 50% of all barcodes in the cell, such that you are sure the main effect in that cell is due to the intended guide. This filtering can easily be done in pandas:
[35]:
filtered_merged_df = merged_df.query("barcode_count_0/barcode_count>0.5")
f"Kept {len(filtered_merged_df):,} / {len(merged_df):,} cells"
[35]:
'Kept 2,146 / 2,896 cells'
Now lets visualize some important stats that allows you to gauge the reliability of the data.
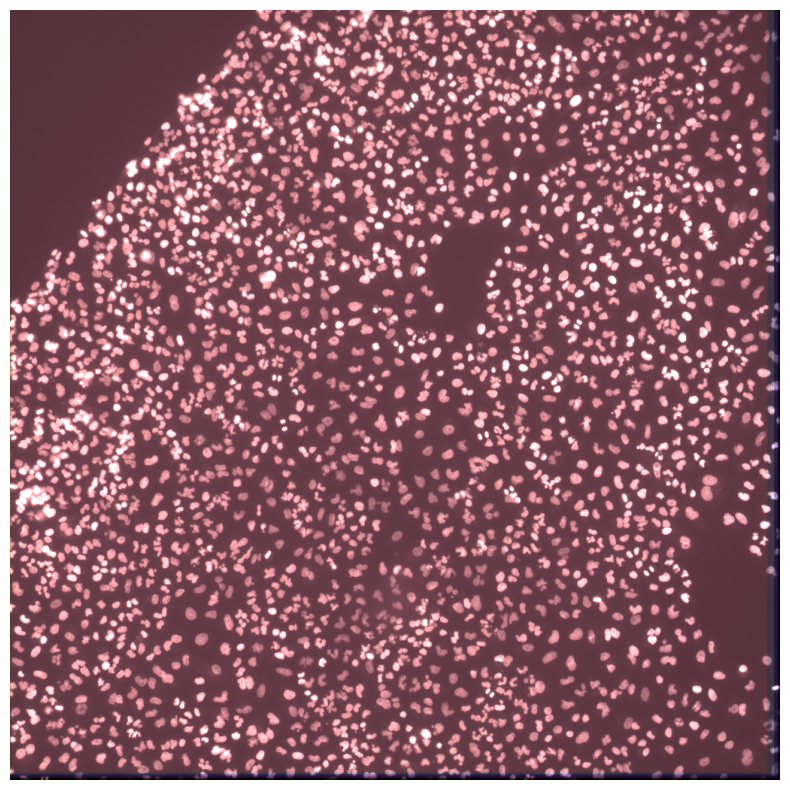
Percent of cells that contain barcode reads
We might want to know how many exact matches are respresented in cells. We can do this by using in_situ_barcode_hist_plot on the df_reads dataframe we computed earlier:
[36]:
import matplotlib.pyplot as plt
from scallops.visualize.histogram import in_situ_barcode_hist_plot
in_situ_barcode_hist_plot(df_reads);
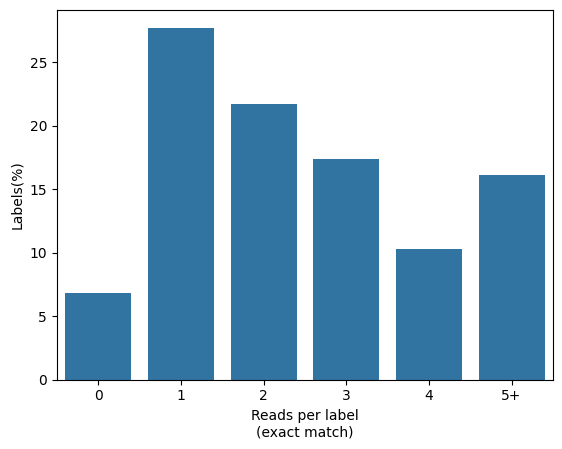
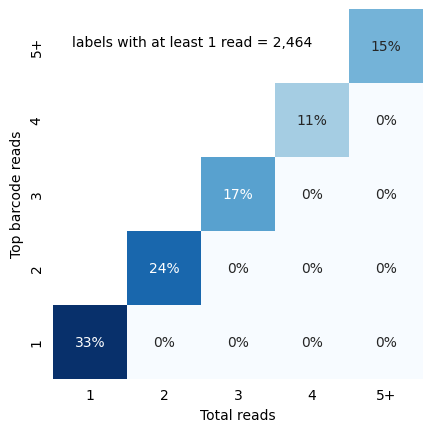
Cellular read distribution categorized by read identity
To visualize how the top barcode reads match the total number of reads, we can use the in_situ_identity_matrix_plot in the df_cells dataframe computed earlier:
[37]:
from scallops.visualize.heatmap import in_situ_identity_matrix_plot
in_situ_identity_matrix_plot(df_cells.rename(columns={"barcode_0": "barcode"}));
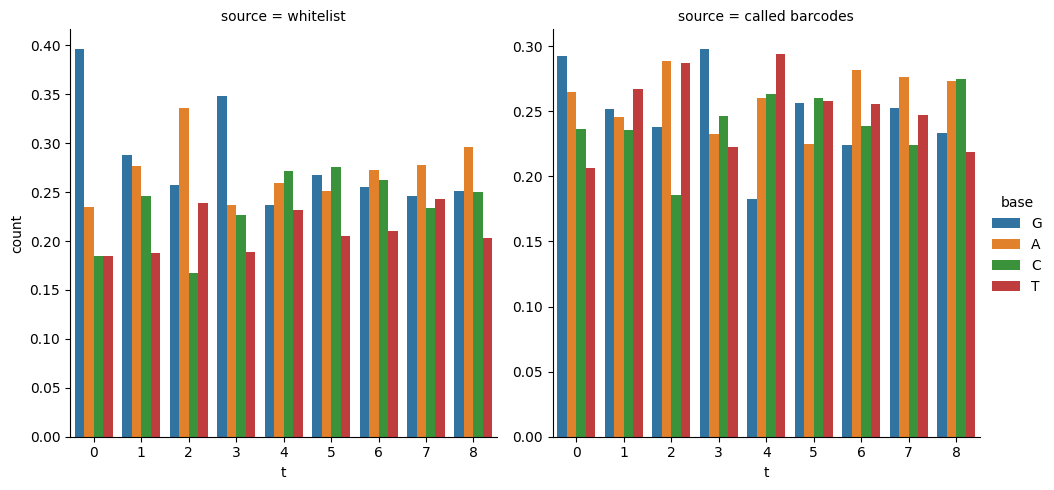
Whitelist and Called Barcodes Distribution
Once you have the called reads, you might want to compare them with your white list, especially in composition. We can use pandas and seaborn to that end:
[38]:
import seaborn as sns
from scallops.reads import base_counts
barcode_counts_df = base_counts(
df_barcode, normalize=True
) # compute counts per base per cycle
barcode_counts_df["source"] = "whitelist"
called_barcode_counts_df = base_counts(df_reads, normalize=True)
called_barcode_counts_df["source"] = "called barcodes"
combined_counts = pd.concat((barcode_counts_df, called_barcode_counts_df))
sns.catplot(
combined_counts,
x="t",
y="count",
col="source",
hue="base",
kind="bar",
sharey=False,
);
Fraction of neighbors with same target
You might also be wondering what is the proportion of neighbors have the same guide. This might be interesting to see the potential biases in guide uptake, and to see how uniform the experiment is. We can achieve this with scipy’s KDTree to find nearest neighbors based on coordinates of the filtered dataframe:
[39]:
from scipy.spatial import KDTree
kdt = KDTree(
filtered_merged_df[["AreaShape_Center_Y", "AreaShape_Center_X"]].values
) # Create a K dimensional tree
neighbors = kdt.query_ball_tree(
kdt, r=40
) # Find all pairs of points between self and other whose distance is at most r.
neighbors_df = filtered_merged_df[["gene_symbol"]].copy()
fraction_same_gene = []
number_of_neighbors = []
for i in range(len(neighbors)):
indices = neighbors[i]
n = len(indices) - 1
target_gene = neighbors_df["gene_symbol"].values[i]
n_same = (neighbors_df["gene_symbol"].values[indices] == target_gene).sum() - 1
fraction_same_gene.append(n_same / n if n > 0 else 0)
number_of_neighbors.append(n)
neighbors_df["number_of_neighbors"] = number_of_neighbors
neighbors_df["fraction_same_gene"] = fraction_same_gene
neighbors_df = neighbors_df.sort_values("fraction_same_gene", ascending=False)
neighbors_df
[39]:
| gene_symbol | number_of_neighbors | fraction_same_gene | |
|---|---|---|---|
| 2631 | non-targeting | 8 | 0.875000 |
| 1777 | non-targeting | 7 | 0.857143 |
| 1196 | non-targeting | 6 | 0.833333 |
| 1645 | non-targeting | 11 | 0.727273 |
| 1941 | non-targeting | 10 | 0.700000 |
| ... | ... | ... | ... |
| 2301 | NaN | 3 | -0.333333 |
| 359 | NaN | 2 | -0.500000 |
| 2878 | NaN | 2 | -0.500000 |
| 867 | NaN | 2 | -0.500000 |
| 2826 | NaN | 1 | -1.000000 |
2146 rows × 3 columns